
Die Validierung hochintegrierter Computersysteme, wie beispielsweise BAAN oder SAP R/3, stellt vollkommen neue Anforderungen an die Verantwortlichen. Die gesetzlichen Grundlagen der Computervalidierung beinhalten überwiegend formale Anforderungen, Definitionen und Vorgehensmodelle. Um den Aufwand aus Sicht des Anwenders in Grenzen zu halten, aber dennoch das Ziel eines gesicherten Systembetriebs zu erfüllen, sind eine Reihe sogenannter kritischer Erfolgsfaktoren zu beachten.
Dr. Rudi Herterich
Anzeige

Die Überwachung von Pumpen in industriellen Prozessen ist weit mehr als eine reine Schutzmaßnahme für das Pumpenaggregat. Neben der präventiven Wartung und...
Die gesetzlichen Anforderungen der Computervalidierung werden von drei unabhängigen nationalen und internationalen Institutionen festgelegt, der FDA (Food and Drug Administration – US), der EU (European Union – EU) und des BM für Jugend, Familie und Gesundheit (Germany). Aufsichtsbehörden in Deutschland zur Überprüfung der Einhaltung gesetzlicher Anforderungen sind die föderal unabhängig voneinander eingerichteten Regierungspräsidien, die ohne länderübergreifend koordiniertes Vorgehen agieren. Der Kontrollrhythmus ist unregelmäßig mit bis zu zweimonatiger Vorankündigung.
Der allgemeine, rechtlich verbindliche Rahmen wird durch das BM für Jugend, Familie und Gesundheit in Form der Betriebsverordnung für pharmazeutische Unternehmer (PharmBetrV) gesetzt. Die VO ist seit 01.04.1985 in Kraft. Sie setzt verbindlich die Richtlinien zur ordnungsgemäßen Herstellung und Sicherung der Qualität (Good Manufacturing-Practises-Richtlinie – GMP) sowie die Vorgaben zur Arzneimittelsicherheit der WHO (Weltgesundheitsorganisation) um.
Aufgrund der unterschiedlichen Gesetze und Richtlinien sowie der heterogenen Struktur bei den Kontrollinstitutionen existiert eine hohe Verunsicherung. Eine allgemein anerkannte Vorgehensweise bei der Computervalidierung gibt es nicht. Als Mindestanforderungen gelten jedoch die Prinzipien wie Nachvollziehbarkeit, lückenlose Dokumentation und leichte Zugänglichkeit der Dokumente.
Entwicklung von Computersystemen
Seit Jahren geht der Trend zu integrierten Computersystemen, d.h. die Integration von Stamm- und Bewegungsdaten sowie die Durchgängigkeit von Geschäftsprozessen stehen im Vordergrund. Dazu kommen neue Entwicklungen im Bereich E-Business und Supply Chain Management mit dem Ziel, über Unternehmensgrenzen hinweg Geschäftsprozesse und Datenflüsse zu optimieren. Mit der Internettechnologie und der damit verbundenen Plattformunabhängigkeit wird es möglich, Geschäftsprozesse über System- und damit über Unternehmensgrenzen hinweg zu gestalten. Weiterhin erlauben Portale die anwenderspezifische und applikationsunabhängige Gestaltung von Oberflächen. Nicht mehr die Funktionalität des jeweiligen Software-Produkts, sondern das Ziel der Anwendung steht im Vordergrund. Diese Entwicklungen in der EDV haben starken Einfluss auf die Methoden der Computervalidierung. Klassische Ansätze gehen eher von geschlossenen, eigenentwickelten Systemen wie LIMS, Verwiegung oder PLS aus, die bestimmte GMP-relevante Bereiche eines Unternehmens wie Qualitätssicherung, Fertigung und Labor tangieren. Es gilt nicht mehr nur eine überschaubare Funktionalität zu validieren, sondern mehrere tausend Funktionen, die gegebenenfalls in einer komplexen Beziehung stehen. Nur ein Bruchteil dieser Funktionen ist aber für die Validierung relevant.
Es stellt sich nun die Frage, wie die Prinzipien der Validierung bei der Einführung integrierter Systeme erfüllt werden können, ohne den Aufwand dafür in unbezahlbare Höhen zu treiben. In Projekten gesammelte praktische Erfahrungen, im folgenden als kritische Erfolgsfaktoren dargestellt, helfen dem Anwender bei der Vermeidung der häufigsten Fehler.
Projektorganisation
In vielen Fällen werden die Einführung von Computersystemen und die Validierung des Systems projektorganisatorisch getrennt. Dies verursacht unnötige Kosten, da ein hoher Koordinationsaufwand zwischen den Projekten entsteht, Dokumentationen redundant zu pflegen sind, die unterschiedlichen Arten von Tests nicht koordiniert durchgeführt werden und damit der Nachweis des validierten Systems gegenüber Dritten schwer darstellbar ist.
Die Validierung muss Bestandteil des Einführungsprojekts sein. Die aufbauorganisatorische Projektgestaltung hängt von individuellen, projektspezifischen Gegebenheiten ab. Sinnvoll ist es, die Validierung als Service der Projektleitung zu definieren.
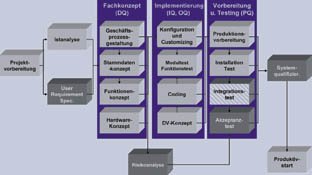
Die Ablauforganisation entspricht der üblicher Vorgehensmodelle zur Einführung integrierter Systeme, erweitert um validierungsspezifische Tätigkeiten, wie in Abbildung 1 dargestellt. Aus der Sicht der Validierung wichtige zusätzliche Aufgaben sind die User Requirement Specification (URS), die Risikoanalyse und die Systemqualifizierung.
Dokumentation
Grundsätzlich müssen alle Dokumente, die bei der Einführung des Computersystems erstellt werden, formal den Anforderungen der Validierung entsprechen. Weiterhin ist darauf zu achten, dass die einzelnen Dokumente über alle Projektphasen hinweg formal und inhaltlich aufeinander aufbauen. Das beginnt bei einfachen Dingen wie der Bezeichnung eines Dokuments und deren Fortführung über alle Projektphasen hinweg bis zur Definition von Begriffen und Sachverhalten. Je strukturierter und logischer die Dokumentation aufgebaut ist, desto einfacher ist letztlich die Handhabung und Pflege im Rahmen des Change Managements.
Prozessstruktur
Die hohe Komplexität integrierter Computersysteme, die funktional nahezu alle Bereiche eines Unternehmens betreffen, erfordert eine anwenderorientierte Sicht auf die Struktur der Dokumentation. Den Mitarbeitern im Unternehmen kann nicht zugemutet werden, sich in die Architektur des Systems einzuarbeiten. Deshalb ist es heute selbstverständlich, die Dokumentation der Validierung prozessorientiert, also entsprechend der Ablauforganisation im Unternehmen aufzubauen. Die Herausforderung besteht allerdings darin, die Prozessstruktur so zu gestalten, dass diese exakt die Tätigkeitsfelder bzw. die Geschäftsvorfälle der Anwender, die von der Validierung betroffen sind, darstellt. Entspricht diese Struktur nicht den ablauforganisatorischen Zusammenhängen, findet die Validierung nicht die notwendige Akzeptanz bei den betroffenen Mitarbeitern im Unternehmen.
Prozessmodelle
Prozessmodelle beschreiben in chronologischer Folge den funktionalen Ablauf einer Tätigkeit, die der Anwender am Computersystem ausführt. Es gibt unterschiedliche Modellierungsmethoden wie Flussdiagramme, Vorgangskettendiagramme (VKD), Swimlanes oder ereignisgesteuerte Prozessketten (EPK). In der Praxis haben sich die EPKs bewährt, da diese weitestgehend frei modellierbar sind. Bei der Modellierung spielt die exakte, genau auf die Validierung fokussierte Beschreibung der Tätigkeit eine entscheidende Rolle. Die Beschreibung der Funktionen in den Modellen sollte auf logischer Ebene erfolgen, damit ist sichergestellt, dass sich diese mit hoher Wahrscheinlichkeit über den gesamten Lebenszyklus nicht ändern und damit keinen Aufwand im Change Management anfällt. Das größte Risiko besteht darin, zu viele unterschiedliche Sachverhalte in die Beschreibung des Prozesses mit einzubringen, was zu Lasten der Verständlichkeit, Übersichtlichkeit und Wartbarkeit geht.
Risikoanalyse
Die Risikoanalyse wird, wie die gesamte Validierung prozessorientiert durchgeführt und sollte unterschiedliche Detaillierungsgrade durchlaufen. Drei Stufen haben sich als sinnvoll herausgestellt, die Modul-, die Stammdaten- und die Prozessebene. Finanzwirtschaftliche Module sind für die GMP-Validierung uninteressant, genauso Stammdaten, die keine qualitätsrelevanten Informationen enthalten. Alle Prozesse, die mit dem als unkritisch identifizierten Modul abgewickelt werden, bzw. alle Prozesse, die ausschließlich auf unkritische Stammdaten zurückgreifen, können damit ausgeschlossen werden. Dadurch reduziert sich die zu untersuchende Anzahl an Prozessen von integrierten Computersystemen erheblich, wie das Abbildung 2 beispielhaft für SAP-R/3 beschreibt. Für die Risikoanalyse der restlichen Prozesse ist die Erstellung eines Fragenkatalogs sinnvoll, der systematisch GMP-relevante Kriterien des Prozesses abfragt.
Die Definition des Begriffs GMP-Risiko ist in den Gesetzestexten und Leitlinien allgemein formuliert. Es ist zu empfehlen, das Risiko auf die Qualität zu beschränken und betriebswirtschaftliche Risiken nicht zu validieren. Im ersten Fall bleibt die Validierung überschaubar, im zweiten Fall sind nahezu alle Prozesse betroffen.
Standard Operation Procedures
Alle Risiken, die sich nicht direkt über das System ausschließen lassen, müssen über Standard Operation Procedures (SOP) abgefangen werden. Dabei handelt es sich um Arbeitsanweisungen, die dem Anwender vorgeben, wie er das System zu bedienen hat. Oft gibt es unterschiedliche Pfade (über Menü, Bottom oder Hotkey), eine Transaktion zu starten. Zur Vermeidung von Risiken, die sich auf unqualifizierte Bedienung des Systems zurückführen lassen, ist es notwendig, die genaue Vorgehensweise am System festzulegen. Dafür bieten sich tabellarische Beschreibungen – sogenannte Quickstarts – an, die exakt die durchzuführenden Schritte am System vorgeben. Der Vorteil besteht darin, dass die Quickstarts für die Erstellung der Testvorlagen wiederverwendet werden können, was den Aufwand deutlich reduziert.
Tests
Entsprechend der prozessorientierten Philosophie müssen sich auch die Tests streng am Geschäftsprozess orientieren. Die Testfälle ergeben sich aus dem identifizierten Risiko. Als Basis für die Testvorlagen dienen die SOPs. Dabei lassen sich drei Arten von Tests unterscheiden. Modultests weisen nach, dass bestimmte isolierte Funktionen in der gewünschten Ausprägung funktionieren. Diese Tests führen die Mitarbeiter in der DV-Abteilung aus.
In den Fachabteilungen durchgeführte Integrationstests stellen sicher, dass globale Geschäftsprozesse wie der Verkauf ausgewählter Artikel und damit für ausgewählte Stammdaten über alle Teilprozesse hinweg sicher funktionieren.
Akzeptanztests beim Anwender sollen garantieren, dass das Customizing und die Pflege der Stammdaten vollständig und abgeschlossen ist. Aus diesem Grunde werden für einen Teilprozess möglichst viele unterschiedliche Stammdaten getestet.
E cav 206
Unsere Webinar-Empfehlung

Der Webcast MTP und modulare Produktion bietet eine einzigartige Gelegenheit, mehr über die aktuellen Entwicklungen bei MTP und in der modularen Produktion zu erfahren.
Chemie- und Pharmaproduktion braucht mehr Flexibilität
In der heutigen sich schnell wandelnden Welt stehen…
Teilen: