Für die Herstellung steriler Produkte und Arzneimittel gelten besondere Anforderungen, um das Risiko einer Kontamination mit Mikroorganismen und Partikeln oder einer Kreuzkontamination mit einem anderen Produkt möglichst gering zu halten. Vieles hängt von der Validierung von Autoklaven und Sterilisatoren ab, denn die Sterilität der verwendeten Produktionsgegenstände, der Reinraumbekleidung und der Primärverpackungen ist ein wesentlicher Aspekt dieser Arzneimittelherstellung.
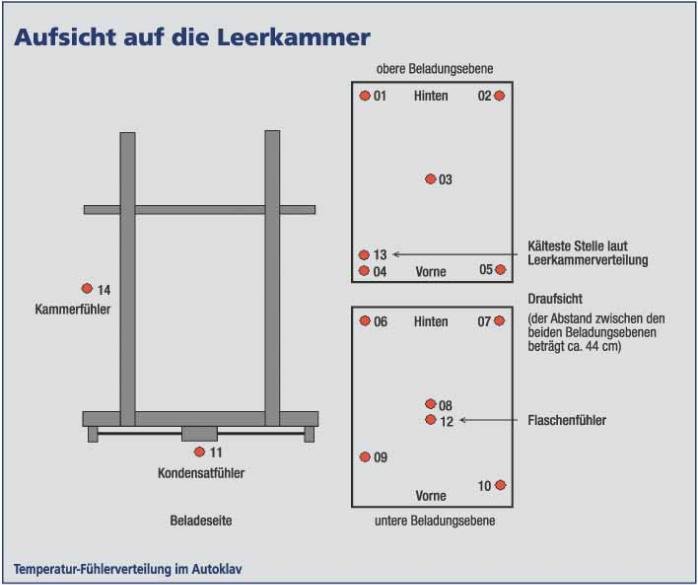
Leerkammerverteilung zur Ermittlung der kältesten Stelle in dem Autoklav mit externen Temperatursensoren und im Kondensatablauf
Zur Reproduktion des Sterilisationsprozesses: Maximalbeladung mit Ampullen
Für die Herstellung steriler Produkte und Arzneimittel gelten besondere Anforderungen, um das Risiko einer Kontamination mit Mikroorganismen und Partikeln oder einer Kreuzkontamination mit einem anderen Produkt möglichst gering zu halten. Vieles hängt von der Validierung von Autoklaven und Sterilisatoren ab, denn die Sterilität der verwendeten Produktionsgegenstände, der Reinraumbekleidung und der Primärverpackungen ist ein wesentlicher Aspekt dieser Arzneimittelherstellung.
Diese Validierung von Ausrüstungsgegenständen mit den einzelnen Herstellverfahren wird von Gesetzgebungen und Richtlinien, wie z.B. der GMP-Leitfaden mit dem Anhang 1, Herstellung steriler Arzneimittel innerhalb der Pharma-Betriebs-Verordnung, das Arzneimittelgesetz als Rechtsgrundlage zur Herstellung von Arzneimittel in Deutschland, der Code of Federal Regulations (CFR) als Gesetzesgrundlage für die amerikanische Kontrollbehörde FDA und die PIC-Richtlinien (Pharmaceutical Inspection Convention) mit gegenseitig anerkannten Empfehlungen, vorgeschrieben.
Alle Sterilisationsverfahren sollten validiert sein. Auf diese kurze Formel des Anhang 1 zum GMP-Leitfaden lässt sich sehr viel Arbeit, Aufwand und Kosten herunterbrechen, denn je nach Anzahl von verschiedenen Sterilisations-Programmen und dazugehörigen Beladungszuständen kann bei der Validierung eines Autoklaven, auch durch ein Muss der dreimaligen Durchführung der Verteilungsstudien mit externen Temperatursensoren und Bioindikatoren, ein sehr hoher Aufwand und Kosten entstehen.
Da auch die Sterilisationszeit bei den verschiedenen Arzneibüchern (Deutsches und Europäisches Arzneibuch, USP 25) unterschiedlich definiert ist, sollten im Vorhinein der Validierung die internen Firmenspezifikationen und Bewertungskriterien (Definition des f0-Wertes, Sterilisierzeit, Einwirkzeit D, usw.) der Validierung eindeutig geklärt sein. Das europäische Arzneibuch beschränkt sich mit der Definition der Sterilisationszeit ?at a minimum of 121 °C for 15 min“, das Deutsche Arzneibuch hat darüber hinaus noch eine Akzeptanz von ± 2 °C und ± 10 kPa am Kaltpunkt für Sterilisationsprozesse spezifiziert. Die amerikanische USP geht außer der Aussage bei der Temperatur ?not less than 121 °C..“ zusätzlich noch auf die f0-Berechnung ein.
Bevor aber die Validierung eines solchen Autoklaven durchgeführt werden kann, muss auch diese Anlage mittels einer Qualifizierung auf Konformität mit den technischen Spezifikationen (DQ) im tatsächlichen Aufbau (IQ), wie verwendeten Materialien, Rohrverbindungen, elektrische und MSR-Komponenten, Medien und Dokumentation untersucht werden. Nach der Kontrolle der Installation ist ein weiterer wesentlicher Gesichtspunkt die normgerechte Kalibrierung aller prozessrelevanten Messstellen. Denn dadurch, dass solche Autoklaven genaue Spezifikationen bei der Sterilisationsphase (121,1 °C ± 0,5 °C) besitzen und die Messkette mit allen Bauteilen mindestens 3-mal so genau arbeiten (± 0,15bis 0,2 °C) soll, ist auch an das Kalibrier-Equipment eine Mindestgenauigkeit von ± 0,05 °C gefordert.
Innerhalb der OQ werden noch die Alarm-, Schalt- und Regelfunktionen sowie eine Leerkammerverteilung zur Ermittlung der kältesten Stelle in dem Autoklav mit externen Temperatursensoren, auch im Kondensatablauf durchgeführt (auch mit Bioindikatoren möglich). Diese externen Temperatursensoren müssen, sofern es sich um Thermoelemente handelt (sind wegen der kurzen Reaktionszeit und der Robustheit gegenüber den trägen und meist empfindlichen PT100-Sensoren zu bevorzugen), vor und nach den Validierungsläufen im eingebauten Zustand kalibriert werden. Die Prekalibrierung sollte bei drei Prüfpunkten um den Arbeitstemperaturbereich herum, die Postkalibrierung bei einem Prüf-/Arbeitspunkt durchgeführt werden. Bei sehr aufwändigen Verteilungsstudien sollte man die Postkalibrierung auch mal zwischen den einzelnen Programm- oder Beladungsänderungen durchführen. Das soll verhindern, dass ? wenn man die Validierung abgeschlossen hat ? sich herausstellt, dass einer oder mehrere Sensoren aus den geforderten Spezifikationen von ± 0,5 °C herausgefallen sind. Bei dem ebenfalls einzusetzenden externen Drucksensor, zur Überwachung und Aufzeichnung der Validierungsstudien mit Lethalitätsberechnung, ist nur die vorherige Kalibrierung erforderlich.
Validierungsplan
Der zu erstellende Validierungsplan (PQ) sollte außer den üblichen Informationen, wie Anlagendaten, Verantwortlichkeiten und Zielsetzung, sowie den mitgeltenden firmenspezifischen SOPs und Dokumente auch valiierungsbezogene Spezifikationen enthalten. Diese sind z. B. Programmarten, Beladungszustände und die Definitionen der kältesten Stellen bzw. der Sterilisationszeit, sowie das Aufführen und die Anzahl der externen Messsensoren (lt. DIN EN 554 für Autoklaven bis 1 m³ entsprechend 12 externen Temperatursensoren und ein Drucksensor; je weiterer m³-Raumvolumen zwei zusätzliche Temperaturfühler) und der Bioindikatoren vom Typ Bacillus stearothermophilus mit einer Keimbelastung von mindestens 106.
Die Verteilung der zu sterilisierenden Beladungsart und Menge (Worst-Case-Szenario), der externen Temperaturfühler mit Bioindikatoren, muss in dem Validierungsplan beschrieben und am besten per einfacher Tabelle und Skizze dargestellt werden.
Zusätzlich sollten per Digitalkamera auch mindestens ein Bild je Beladungszustandund der Fühler- bzw. Indikatorenverteilung erstellt werden, das dann mit in den Bericht und in die Auswertung einfließt.
Bei der Durchführung der drei Validierungsstudien pro Programm und Beladung ist ebenfalls darauf zu achten, dass die Ausgangsbedingungen, wie Anfangstemperatur des Autoklaven und der Beladung, die Beladungsart und -menge, die Fühlerverteilung sowie die Lagerbedingungen der Bioindikatoren (kühle Lagerung) immer gleich sind. Hiervon hängen einfach die reproduzierbaren Ergebnisse der Validierung eines solchen Gerätes ab. Gerade bei der Wärmeabstrahlung einer Worst-Case-Beladung ist dies sehr wichtig, denn die Anfangstemperatur eines Autoklaven und dessen Beladung beim Start von Validierungsläufen sollte sich immer unterhalb von 40 °C befinden. Sonst ist der Verlauf des Sterilisationsprozesses verfälscht und für Validierungsdokumentationen nicht verwendbar ? der Sterilisationsprozess verläuft nicht reproduzierbar ab. Dies ist aber leider durch die vorhandene Masse bei Maximalbeladung auch nur sehr zeitaufwändig umzusetzen.
Auswertung
Nach den Verteilungsstudien werden die Messwerte der externen Sensoren und die hoffentlich nicht vorhandene Wachstumsrate der Bioindikatoren ausgewertet. Die Temperatur- und Druckauswertungen können bei den handelsüblichen und etablierten Validierungsmesssystemen im Vorhinein programmiert und später automatisiert durchgeführt, gespeichert und ausgedruckt werden.
Die Bebrütung der verwendeten Bioindikatoren sollte dann umgehend in einem Wärmeschrank bei einer Temperatur von 56 °C ± 2 °C über mindestens 3 bis 5 Tagen zusammen mit den Positivkontrollindikatoren erfolgen. Der große Unterschied und dadurch auch eine Arbeitserleichterung zwischen den Indikatorenvarianten ist, dass die Indikatorenampullen direkt in den Brutschrank eingelegt und ausgewertet, die Sporenstreifen aber vorher noch in ein Nährmedium gelegt werden müssen.
Nachdem man alle Ergebnisse in dem Bericht eingebunden hat, ist es wichtig, eine zusammenfassende Bewertung der Einzelergebnisse innerhalb der Erst-Validierung pro Beladungs- und Programmart zu erstellen und die zyklische Durchführung der Revalidierungsmaßnahmen zu beschreiben.
Hier reicht es normalerweise aus, wenn man die Revalidierung so strukturiert, dass in einem regelmäßigen jährlichen Zyklus ein bis zwei exemplarische Beladungszustände und Programmarten getestet werden, um in 3 bis 5 Jahren alle möglichen Beladungen und Programme wenigstens einmal geprüft zu haben. Sollten sich während der Zeit die Zustände an der Anlage oder bei den Beladungen ändern, so müssen diese Veränderungen mit einer Ausgangs- oder Erstvalidierung ausführlich getestet werden.
Wolfgang Hähnel
Unsere Webinar-Empfehlung

Der Webcast MTP und modulare Produktion bietet eine einzigartige Gelegenheit, mehr über die aktuellen Entwicklungen bei MTP und in der modularen Produktion zu erfahren.
Chemie- und Pharmaproduktion braucht mehr Flexibilität
In der heutigen sich schnell wandelnden Welt stehen…
Teilen: