Sämtliche Produkte, die in GMP-pflichtigen Bereichen der pharmazeutischen Industrie Einsatz finden sollen, müssen in Übereinstimmung mit den Qua- litätsanforderungen der pharmazeutischen Industrie entwickelt und hergestellt worden sein. Für einen Zulieferer von Software heißt dies: Bereitstellung validierungsfähiger Software, Einhaltung von qualitätssichernden Maßnahmen und Erstellung der zugehörigen Dokumentation.
Autoren: André Kurig, Head of Industry Pharma, CSB-System Martin Miller, Vertrieb, CSB-System
In der pharmazeutischen Industrie werden diverse Planungs-, Produktions- und Dokumentationsprozesse mithilfe computergestützter Systeme realisiert. Die Validierung dieser computergestützten Systeme in GMP-pflichtigen Bereichen ist ein zentrales Element der qualitätssichernden Maßnahmen. Die Thematik umfasst sowohl alle Qualifizierungs- und Validierungsmaßnahmen als auch Aspekte wie Datensicherheit und Datenintegrität bis hin zum Einsatz elektronischer Unterschriften. Im Wareneingang werden Rohstoffe und Packmittel durch Online-Kontrollen mengen-, gewichts- oder volumenmäßig mit Konzentration bzw. Aktivität (z. B. Enzyme) chargenweise im System mit Lagerplatzzuordnung sowie Eingangs- und Verfallsdatum erfasst. Den Wareneingangsstammdaten müssen außerdem Informationen wie Chargennummer des Lieferanten, die interne Wareneingangsnummer und Lieferantenangaben zugewiesen werden. Zum Abschluss der Wareneingangserfassung erhält jede Charge ein Etikett mit einem Unikat-Barcode, der die Charge eindeutig identifiziert und somit die Rückverfolgbarkeit gewährleistet. Die Waren können im System in die entsprechenden Qualitätskontrollstatus („in Quarantäne“, „frei“ oder „gesperrt“) gesetzt werden. Das Umetikettieren der Produkte ist nicht mehr notwendig. Eine branchenspezifische Software wie das CSB-System schlägt unter Berücksichtigung der Parameter, welcher Artikel von welchem Lieferanten eingelagert werden soll, automatisch die spezifischen Prü- fungen vor.
Im Rahmen der durchgängigen Qualitätskontrolle für Rohstoffe, Packmittel, Zwischenprodukte, Bulkware und Endprodukte müssen in der Qualitätskontrolle Proben gezogen und Identifikationsprüfungen durchgeführt werden. Nachdem die Identität der Charge durch eine positive qualitative oder quantitative Analytik bestätigt worden ist, werden diese von den Herstellungs- und Kontrollleitern gemeinsam freigegeben. Zugehörige Qualitätsparameter, wie beispielsweise pH-Wert, Konzentration, Aktivität usw., können im System hinterlegt werden. Das CSB-System überwacht außerdem die Verfallsdaten.
Ausgangspunkt der Produktion ist die Herstellvorschrift, die die stoffliche Rezeptur sowie die zugehörigen technischen Verfahren beschreibt. Zur Vorbereitung von Aufträgen auf Basis einer freigegebenen Herstellvorschrift rechnet die Software die Rezepturmenge auf die tatsächliche Auftragsmenge – unter Berücksichtigung von Gehalt bzw. Aktivität der bei der Qualitätsprüfung bestimmten Werte – hoch. Anschließend werden entweder Auftragsunterlagen wie Ansatzrezepturen und Lagerbereitstellungslisten auf Papier erstellt oder online abgearbeitet. Während der Auftragsdurchführung hat die Arbeitsvorbereitung stets die Möglichkeit, laufende Aufträge und deren Stand in der Produktion zu überwachen. Alle Abweichungen werden angezeigt und protokolliert. Die online erfassten Auftragsdaten können jederzeit als Protokolle ausgegeben oder archiviert werden. Zudem wird über die Daten eine lückenlose Chargenrückverfolgung gewährleistet.
Der Benutzer wird mittels Workflow-Unterstützung durch die einzelnen Arbeitsschritte des gesamten Herstellungsprozesses geführt. Die in der pharmazeutischen Herstellung benötigten Wägefunktionen wie Mengenumrechnung, Austauschstoff, Dichteberechnung und Bruttowägung stehen systemgestützt zur Verfügung.
Während der Auftragsdurchführung kann der Mitarbeiter auf die Sicherheitsvorschriften zum Arbeits- und Brandschutz online zugreifen. Das CSB-System trägt somit zur Vereinfachung und Unterstützung bei allen systemgeführten Herstellschritten bei. Als weitere Sicherungsmaßnahme ist eine Barcode-Identifikation zur Online-Kontrolle von Einsatzstoffen möglich. Neben der Kontrolle der verwogenen Ausgangsstoffe und der Erfassung der zudosierten Rohstoffe kann die Herstellvorschrift konsequent – entweder benutzergeführt durch Auslösen von SPS-gesteuerten Prozessschritten oder durch Einbinden in Prozessleitsysteme – abgearbeitet werden. Nach dem Erfassen von eventuellem Ausschuss erfolgen die Auftragsfertigmeldung und der Ausdruck von Barcode-Etiketten mit variablen Layouts für die Verpackungseinheiten.
Systemsicherheit
Das System registriert und dokumentiert für jede hergestellte Charge, welcher Mitarbeiter wo, was, wie, wann und mit welchen Artikeln (Rohstoffe, Ausgangsstoffe, Bulkware, Packmittel) durchgeführt hat. Neben der bereits erwähnten Archivierung der Herstelldaten – entsprechend den gesetzlichen Vorgaben bis zu 30 Jahre – wird das Ausfallrisiko des Systems durch Sicherheitskonzepte minimiert, beispielsweise durch gespiegelte Systeme und mitlaufende Logfiles.
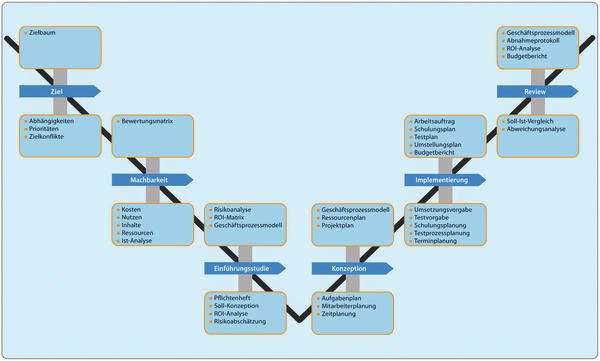
Prospektive Systemvalidierung
Bevor eine IT-Lösung Aufgaben in der pharmazeutischen Herstellung übernehmen darf, muss das System im Rahmen der pharmazeutischen Qualitätssicherung prospektiv, d. h. vorausschauend, validiert werden. Mit dem Abkommen zur gegenseitigen Anerkennung von GMP-Inspektionen zwischen der FDA und der EU wird die in den USA zu beobachtende Tendenz, sich bei FDA-Inspektionen verstärkt auf die eingesetzten Computersysteme (Hard- und Software) zu konzentrieren, sicherlich Auswirkungen auf den europäischen Raum haben.
Die gesetzliche Verantwortung zur Validierung computergestützter Systeme liegt beim pharmazeutischen Unternehmen. Aufgrund der Komplexität der Systeme und der meist fehlenden Möglichkeiten, alle notwendigen Tests am Endsystem durchzuführen, ist eine enge Zusammenarbeit mit dem Software-Lieferanten notwendig.
Mittlerweile geschieht dies nach festgelegten Regeln, die sich aus bewährten Vorgehensweisen in Arbeitsgruppen aus Anwendern und Lieferanten solcher Systeme herauskristallisiert haben. Dazu wird entlang eines Lastenheftes und des daraus resultierenden Pflichtenheftes die Software entwickelt, die Hardware qualifiziert und mit festgelegten Testverfahren geprüft.
Erst wenn die tatsächliche Systemfunktion und -leistung den genannten Anforderungen und Spezifikationen in vollem Umfang entspricht und somit dokumentiert ist, gibt der Betreiber das System frei. Da dieser letztlich gegenüber den Überwachungsbehörden verantwortlich ist, kann der Systemlieferant diese Freigabe nur vorbereiten.
Online-Info www.pharmaproduktion.com/0210448
Unsere Whitepaper-Empfehlung

Absicherung der IT-Infrastruktur bei der Lebensmittelindustrie und im Lebensmittelhandel unter KRITIS-Vorgaben: Fakten und Lösungen
Teilen: